Regulatory Submissions with Real-World Evidence: Successes, Challenges, and Lessons Learned
On September 21st, I attended the 𝗗𝘂𝗸𝗲-𝗠𝗮𝗿𝗴𝗼𝗹𝗶𝘀 𝗜𝗻𝘀𝘁𝗶𝘁𝘂𝘁𝗲 𝗳𝗼𝗿 𝗛𝗲𝗮𝗹𝘁𝗵 𝗣𝗼𝗹𝗶𝗰𝘆 𝗮𝗻𝗱 𝗙𝗗𝗔’𝘀 𝗵𝘆𝗯𝗿𝗶𝗱 𝗽𝘂𝗯𝗹𝗶𝗰 𝗺𝗲𝗲𝘁𝗶𝗻𝗴: 𝘙𝘦𝘨𝘶𝘭𝘢𝘵𝘰𝘳𝘺 𝘚𝘶𝘣𝘮𝘪𝘴𝘴𝘪𝘰𝘯𝘴 𝘸𝘪𝘵𝘩 𝘙𝘦𝘢𝘭-𝘞𝘰𝘳𝘭𝘥 𝘌𝘷𝘪𝘥𝘦𝘯𝘤𝘦: 𝘚𝘶𝘤𝘤𝘦𝘴𝘴𝘦𝘴, 𝘊𝘩𝘢𝘭𝘭𝘦𝘯𝘨𝘦𝘴, 𝘢𝘯𝘥 𝘓𝘦𝘴𝘴𝘰𝘯𝘴 𝘓𝘦𝘢𝘳𝘯𝘦𝘥. The first part of this blog was posted on Orizaba Solution’s linked in page on 9/28, skip to the data and/or afternoon session to see information about the medical products and to hear about the afternoon session.
The meeting opened with remarks from Dr. Sara Brenner, FDA Principal Deputy Commissioner, followed by updates on PDUFA VII and MDUFA V commitments. During the PDUFA VII updates, three main reasons were given for rejecting applications in the 𝗔𝗱𝘃𝗮𝗻𝗰𝗶𝗻𝗴 𝗥𝗲𝗮𝗹-𝗪𝗼𝗿𝗹𝗱 𝗘𝘃𝗶𝗱𝗲𝗻𝗰𝗲 𝗣𝗿𝗼𝗴𝗿𝗮𝗺 (𝗔𝗥𝗪𝗘):
1. Study interpretability concerns
2. Study role in the development program
3. Better suited for established pathway engagement
This resonated with my experience at FDA (2019–2023) when working on data standards for RWE submissions. In the submission reviewed, a randomized controlled study was used for the treatment arm and an RWE study for the comparator. In most of the studies, reviewers had determined the 2 cohorts were not comparable, making analyzing patient-level data for safety and efficacy unnecessary. Despite this, sponsors had often formatted the RWE data into CDISC, but that’s another post😊.
The heart of the meeting was the review of 𝗳𝗼𝘂𝗿 𝗰𝗮𝘀𝗲 𝘀𝘁𝘂𝗱𝗶𝗲𝘀 where RWE was submitted as part of the “adequate and well-controlled study” (AWCS) required for approval under 21 CFR 314.126. (Some may remember Dr. John Concato, former Director of OMP, quoting this section of the CFR so often when referring to RWE questions that he felt compelled to note the agency did not require him to memorize it). Each case study was presented from two perspectives: the sponsor's and the FDA's. FDA presentations focused on 𝘁𝗵𝗿𝗲𝗲 𝗸𝗲𝘆 𝗰𝗼𝗻𝘀𝗶𝗱𝗲𝗿𝗮𝘁𝗶𝗼𝗻𝘀 𝘂𝘀𝗲𝗱 𝗶𝗻 𝘁𝗵𝗲 𝗮𝗽𝗽𝗿𝗼𝗮𝗰𝗵 𝘁𝗼 𝗲𝘃𝗮𝗹𝘂𝗮𝘁𝗶𝗻𝗴 𝗥𝗪𝗘:
🔹Are the RWD fit for use?
🔹Does the study design provide adequate scientific evidence for the regulatory question?
🔹Was the study conducted in line with FDA requirements?
👉 Below are the five takeaways relevant to submission of RWE as part of the AWCS.
𝟱 𝗧𝗮𝗸𝗲𝗮𝘄𝗮𝘆𝘀 𝗳𝗿𝗼𝗺 𝗙𝗗𝗔 𝗖𝗮𝘀𝗲 𝗦𝘁𝘂𝗱𝗶𝗲𝘀
1. Ensure treated and comparator patient cohorts are comparable, especially in non-randomized designs where RWE is used in the comparator arm.
2. Ensure the RWD source provides reliable capture of the primary endpoint for all patients.
3. Select endpoints that are objective and measurable; consider independent adjudication where appropriate.
4. Engage early with FDA on interpretability, endpoints, and statistical approaches.
5. CDER and CBER still requires access to patient-level data for approval of a new drug or indication (CDRH does not)—if unavailable, an established regulatory pathway is likely more appropriate.
Case Studies: Medical Product Details
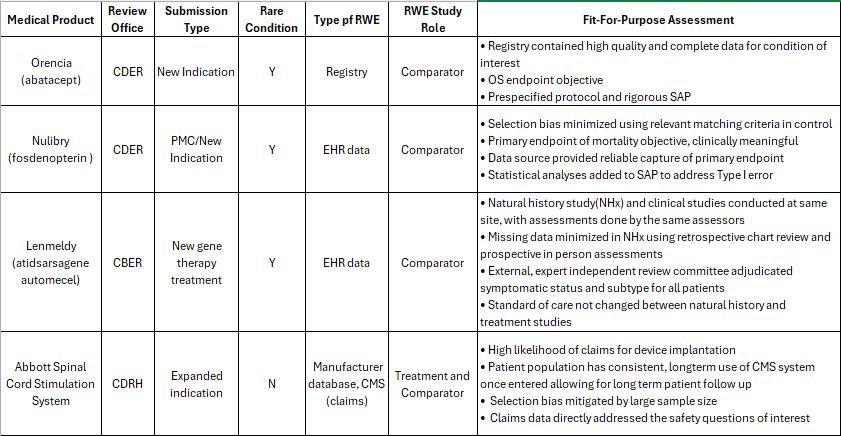
The four medical products using RWE as part of the submission and discussed in the morning sessions.
Exploring the Strengths, Challenges, and Future of Real-World Evidence at FDA
After lunch, Khair ElZarrad, Director of the Office of Medical Policy (OMP), opened the day’s first panel: Strengths and Challenges of Using RWE in Case Studies. This session brought together FDA experts and industry speakers to reflect on lessons learned from the 4 case studies presented at the morning session.
Yun Lu, Deputy Division Director in the Division of Analytics and Benefit-Risk Assessment (DABRA), highlighted several themes from the morning’s case studies:
Three of four applications were for rare diseases.
Registry data played a key role in two cases.
Objective outcomes were selected as primary endpoints.
· Three of the four applications used RWE as external control for a RCT with the treatment.
natural history of diseases, whether used in the approval or not, allowed for an understanding and predictability of disease course important for study design and minimizing bias
Sponsors in all 4 case studies actively addressed bias in the RWE used.
All had early and frequent engagement with FDA and this helped shape their applications
Careful data evaluation, bias minimization, and robust statistical analysis on the part of the sponsors underpinned success.
Motiur Rahman (OMP) added that while sponsors often ask for a “checklist” for RWE, there is no universal template. As former OMP head Dr. John Concato frequently answered when he was asked specific questions about RWD sources, : “it depends.” What matters is whether a study meets the standard of an adequate and well-controlled study (AWCS), as described in 21 CFR 314.126 and this will likely depend on the details of your study design.
When asked about key decision points for using RWE, Dr. Yun emphasized two guiding principles: reliability and relevance.
Reliability means data must be accurate, complete, and traceable.
Relevance means the data must capture critical outcomes and confounders, and include enough patients to power the study.
This explains why a one-size-fits-all checklist doesn’t exist. For example, the CIBMTR registry used in the Orencia approval included every U.S. allogeneic transplant with detailed demographics, making it highly reliable and relevant. In contrast, voluntary registries may be incomplete and unsuitable for certain endpoints. Yun closed with a reminder: RWE must be thoughtfully designed to measure outcomes that truly matter.
Fireside Chat: Envisioning the Future of RWE
The day concluded with a forward-looking discussion moderated by former FDA Commissioner Mark McClellan, now Director of the Duke-Margolis Center for Health Policy. Panelists shared their vision for the next chapter of RWE at FDA:
Dr. Marie Bradley (OMP): Expect more integration and data linkage to create comprehensive datasets and prepare for the growing volume of RWE trials.
Dr. Mallika Mundkur (Office of the Commissioner): RWE and AI remain top priorities, especially through collaboration with NIH, CMS, and other agencies.
Dr. Shantanu Nundy (FDA AI Advisor): Shared a personal story illustrating the value of registries in clinical decision-making. He noted that using AI to look across hospital records to answer questions both clinicians and patients might have would be useful. I would argue putting money into registries is likely to yield better results than AI, at least based on the evidence we have so far.
Dr. Daniel Caños (CDRH): Highlighted the promise of unique device identifiers to connect data sources and improve evidence for devices.
Panelists acknowledged ongoing challenges—interoperability, quality, and traceability—but expressed optimism. They pointed to new initiatives such as Sentinel 3.0 and CDRH’s collaboration on the National Evaluation System for Health Technology (NEST) as steps toward stronger evidence generation and better regulatory decision-making.
Takeaway:
The discussions underscored both the progress and complexity of integrating real-world evidence into regulatory science. While there’s no single roadmap, the principles of reliability, relevance, and early FDA engagement continue to guide successful RWE applications. Looking ahead, greater data connectivity, insight and analysis from cross-agency collaborations, and innovative infrastructure projects promise to expand the role of RWE in shaping the future of drug and device development.