Real-world Evidence and External Control Arms in Oncology: Progress, Friction, and the Path Forward
Introduction
On April 7, 2026, the Friends of Cancer Research (FOCR) convened a public meeting: Application of External Control Arms (ECAs) in Oncology Drug Development. The meeting presented preliminary results from FOCR’s ECA Pilot Project and explored the broader role of real-world evidence (RWE) and ECAs in regulatory decision-making through a keynote session with Dr. Amy Abernathy and 3 panel discussions.
Progress is undeniable. The FDA has now cited 13 approvals across CDER and CBER where RWE was used in regulatory decision-making. But despite this momentum, ECAs, and RWE more broadly, remain a work in progress: scientifically, operationally, and regulatorily.
The meeting itself reflected that tension: optimism about what is possible, paired with a clear acknowledgment of what still needs to be solved.
The Expanding Maturity of RWE
Since the 21st Century Cures Act (2016) and FOCR’s early pilot efforts (2017), the RWE ecosystem has evolved significantly. Data sources have expanded beyond claims to include electronic health records, linked datasets, and patient-reported outcomes. Analytical methods have matured, and the FDA has become more comfortable engaging with these approaches and has published over a dozen guidance documents addressing the use of RWE.
Dr. Amy Abernathy highlighted the progress made with data: “we are now moving toward longitudinal, multimodal data that captures the full patient journey across systems and time.”
However, she noted six key gaps:
Data curation and transparent transformation
AI governance and continuous validation
Scientific rigor and clear statistical plans
Documentation and traceability
Systematic handling of missing data
Convergence toward best practices
In summary, we have more data, better tools, and an understanding of the gaps, but we are still building the collective experience needed for scaling and standardization.
The FOCR ECA Pilot: Defining “Fit for Purpose”
FOCR’s ECA Pilot Project is the fourth in a series of initiatives aimed at developing and establishing methodology for using RWD in regulatory contexts. This pilot aimed to test whether multiple data partners could independently construct an external control arm that replicates a Phase 3 randomized controlled trial (RCT) in metastatic pancreatic cancer.
Selected data partners were given the trial eligibility criteria translated for RWD and asked to construct a cohort independently using their own data source. These cohorts were then compared to the original trial control arm.
The results were encouraging and showed that independent construction of ECAs across heterogeneous data sources is feasible under a shared statistical analysis plan.
At the same time, variability across partners was expected and observed. Differences from the randomized trial were attributed in part to known issues, such as trials enrolling healthier patients than those seen in real-world settings. Reasons for differences will be explored further in the pilot.
The takeaway is nuanced:
Feasibility has been demonstrated, but consistency and reproducibility remain open questions.
FDA Perspective: Fitness-for-Use Is Multidimensional
In the second panel, Dr. Marie Bradley, Senior Advisor for RWE in the Office of Medical Policy at CDER, was asked directly about how sponsors should think about factors such as relevance and completeness when assessing real-world data sources for constructing an ECA.
Dr. Bradley emphasized that fitness-for-use is inherently multidimensional.
At its core is data relevance:
Does the dataset reflect the appropriate patient population?
Does it capture the clinical context and sufficient follow-up?
But relevance alone is not sufficient. Sponsors must also demonstrate:
Clinically meaningful endpoints
Comparability to the trial population
Reliable and consistent measurements for key variables
Notably, the FDA has not defined fixed thresholds for acceptable data quality. Expectations remain context-dependent and evolving.
Another important signal: the FDA continues to require patient-level data submissions for CDER and CBER, even when RWE is used. This is significant because late last year the Agency issued a statement that it was considering allowing RWE submissions to CBER and CDER without requiring identifiable patient-level data in all cases. If implemented, this represents a significant departure from standard FDA submission requirements. Dr. Bradley’s specific mention of this may indicate that the FDA has reconsidered this and will continue with the current requirements.
The Role of Data Standards
One notable gap in the discussion was the role of data standards.
Unlike Sentinel, which relies on a common data model and standardized, centralized query construction, the ECA pilot allowed each partner to independently implement eligibility criteria across heterogeneous data sources.
This raises critical questions:
How much variability is introduced through independent interpretation and what contributes to the variability?
What is gained or lost when data are standardized versus left in native persistent formats?
In cases where cohorts are being created from large data sources, where in the pipeline is the optimal place for standardization to occur?
As ECAs scale and use cases widen, these questions will become increasingly important. As noted in one of the panels, very large datasets are often reduced dramatically when eligibility criteria are applied to create cohorts. I’ve seen patient counts go from the tens of thousands to below 100 after the application of inclusion/exclusion criteria. Standardization to facilitate characterizing the data, for example, the reason for a missing data element, a major issue with RWE, could have significant benefits when analyzing fitness for use.
Standardization may not be required to demonstrate feasibility, but it may be essential for reproducibility, comparability, and operational efficiency.
The Tension: Flexibility vs Certainty
A central theme throughout the meeting was a familiar tension:
Industry wants:
Clear thresholds
Defined benchmarks
Predictability
FDA provides:
Principles
Contextual evaluation
Flexibility as RWE data and methods evolve
As noted during the discussion, stakeholders often want both certainty and flexibility, but the two are inherently in tension.
In an evolving field, this may be unavoidable. Premature standardization could constrain innovation, while insufficient guidance can slow adoption. However, it was underscored that flexibility does not mean the FDA will reduce its evidentiary standards.
Toward a New Evidence Paradigm
Perhaps the most forward-looking insight was this:
We are still framing the conversation as RWD versus randomized controlled trials.
That framing is increasingly outdated.
The future lies in integrated evidence generation. Dr. Abernathy emphasized in her keynote that industry will be able to move faster when we bring RCT and RWE data together and start thinking about the complete evidence package and multiple ways to get there.
One particularly compelling idea raised:
When considering using an ECA, don’t just optimize real-world data to match trials, but optimize trials to better reflect the real world.
This shift could reduce the structural mismatch between trial populations and real-world patients, improving both generalizability and the viability of ECAs.
Conclusion: Progress and the Goal
The promise of RWE is clear:
It offers a pathway to faster, more efficient drug development and the potential to bring therapies to patients sooner. ECAs are one of those pathways.
But the path forward requires balance.
Industry needs to invest in rigorous, transparent, and reproducible methods
Regulators should continue to provide guidance while preserving flexibility in an evolving landscape
Both must resist prematurely regulating what is not yet fully understood in the name consistency for predictability
The goal is not simply to make ECAs work. It is to make them credible, scalable, and decision-grade so we can effectively help bring safe and effective drugs to patients faster.
Implications for Data Strategy and Implementation
At Orizaba Solutions, this is exactly where we focus, helping federal agencies and sponsor companies bridge the gap between data availability and decision-grade evidence. Our work centers on modernizing, integrating, and operationalizing real-world data through data standards, governance, and AI-ready pipelines, with deep experience supporting FDA submissions, understanding RWE submission requirements, and large-scale healthcare datasets. As the use of RWE continues to evolve, we are particularly interested in helping organizations design data strategies and architectures that are not only fit-for-purpose but also scalable, transparent, and submission-ready.
PHUSE US Connect 2026: AI and the Human Factor
I had a great time in Austin at PHUSE US Connect 2026, March 23-26. It was wonderful reconnecting with long-time colleagues, meeting new people, and seeing how the clinical data science ecosystem continues to evolve.
A few themes stood out across the conference.
1. AI is reshaping clinical data workflows.
Not surprisingly, AI was everywhere. Sessions explored how large language models and agent-based systems are being applied to statistical programming, automated dataset generation (SDTM/ADaM), and orchestration of end-to-end analytics workflows.
2. Automation through metadata and standards.
A second major theme was the automation of the clinical data lifecycle. Many talks focused on metadata-driven pipelines that move from protocol to analysis outputs with far less manual intervention.
3. Modernizing data standards and interoperability.
There was significant discussion around CDISC modernization, biomedical concepts, and initiatives like CDISC 360i, as well as the growing importance of interoperability with standards such as FHIR.
4. Real-World Evidence continues to gain momentum.
Real-world data and evidence had its own dedicated stream this year. Sessions addressed RWD quality, external control arms, and integration with clinical trial data.
I was also pleased that Orizaba Solutions presented work in this area focused on standards for submitting data derived from real-world sources to regulators. I’ll share that presentation soon.
Two keynotes that emphasized the human side of data
Despite the focus on AI, the keynote speakers highlighted something equally important: how humans interpret and communicate data.
Lynne Peeples opened the conference with a talk titled “When Data Doesn’t Speak for Itself.” She reminded us that biomedical data is often assumed to be objective and self-explanatory. The COVID-19 pandemic showed how easily data can be misunderstood or mistrusted when uncertainty and context are not communicated clearly.
She introduced a helpful framework for communicating data using the acronym CLEAR:
Context – Provide the background needed to interpret the data
Limitations – Be transparent about what the data cannot show
Explain – Clarify who collected and analyzed the data
Audience – Meet people where they are
Representation – Be thoughtful about how the data is presented
It’s a powerful reminder that good data science also requires good communication.
The closing keynote from Dr. Lilliam Rosario (TransCelerate) focused on how data standards enable collaboration and better regulatory decision-making.
Her message resonated strongly:
Standards give us a shared language that allows the ecosystem to learn together.
Structured clinical data allows knowledge to accumulate across programs, improving the fidelity of risk-benefit decisions and enabling new insights. Initiatives in toxicology data sharing and clinical trial data sharing are already demonstrating how harmonized data can unlock cross-study learning.
One idea she emphasized stuck with me:
Digitalization is not modernization. It’s risk mitigation.
When sponsors, solution providers, and regulators align on structured data exchange, safety insights can move through the system rather than being reconstructed again and again.
Final reflection
If there was one underlying message from PHUSE this year, it’s this:
AI may accelerate analysis, but structured data and shared standards are what make learning possible.
As the industry continues to expand the use of real-world data and AI-driven analytics, ensuring that data is well-structured, traceable, and fit for regulatory use will only become more important.
This is exactly the type of challenge we enjoy working on at Orizaba Solutions.
Looking forward to continuing these conversations with colleagues across the community.
CDISC U.S. Interchange 2025: Connecting Standards, Strategy, and the Future of Clinical Research

CDISC CEO and President, Chris Decker, kicking off the CISC U.S. Interchange 2025 in Nashville, TN.
At Orizaba Solutions, we believe data is only as powerful as the strategy and standards behind it. That’s why the CDISC U.S. Interchange 2025, held in Nashville this October, felt like both a reunion and a reminder of how far the data standards community has come.
I’ve attended CDISC U.S. Interchanges periodically since my first in 2005, when I had just transitioned from the genomics world and was astonished by how differently clinical trial data was being managed. The last Interchange I attended was in 2019, when Real-World Data (RWD) first appeared on the agenda, alongside early discussions about CDISC 360 and the CDISC Library.
Much has changed since then. The world, and CDISC, has evolved significantly under new leadership, now guided by Chris Decker as CEO. The 2025 Interchange brought together thought leaders, innovators, and data standards experts from across the clinical research ecosystem to explore the future of connected, standards-driven science.
Over two days, sessions covered digital study design, regulatory submissions, TMF modernization, AI-driven automation, and real-world data. The program reflected CDISC’s 25-year legacy of bringing together a global community of experts to develop and advance data standards of the highest quality to create clarity in clinical research.
The opening session honored pioneers who built the foundation for today’s standards and were instrumental in supporting their adoption, including Dr. Lilliam Rosario, former FDA Director of the Office of Computational Science, and Stephen Wilson, former USPHS Director of the Division of Biometrics III, among others from FDA and industry whose work and championship has shaped the submission data landscape.
Keynote: Creating an AI-Enabled Learning Health and Research System — Now It’s Personal
One of the most powerful sessions was the keynote by Dr. Peter J. Embí, Professor of Biomedical Informatics and Medicine at Vanderbilt University and Founding Director of the ADVANCE AI Center. His presentation, “Creating an AI-Enabled Learning Health and Research System — Now It’s Personal,” combined personal experience with a vision for transforming healthcare.
Dr. Embí recounted his years-long struggle with misdiagnosis before finally learning he had a rare adrenal cancer—an experience that exposed the deep challenges of diagnostic accuracy and data fragmentation. Citing the Institute of Medicine’s report To Err Is Human (2000) and other research, he reminded the audience that diagnostic errors remain one of the most under-addressed threats to patient safety. “People are suffering needlessly,” he said. “We can and must do better.”
Central to his message was the concept of the Learning Health System (LHS)—an adaptive framework where data from every patient encounter informs continuous improvement in care, research, and policy. The traditional separation between research and clinical care, he noted, has created “silos of excellence” that hinder progress. Local learning systems, designed to “learn from every patient,” can bridge those divides.
While AI has immense potential to accelerate this transformation, Dr. Embí cautioned that “AI is not magic—it is only as good as the data it is trained on.” He warned of algorithmic bias, citing an example where healthcare costs were used as a proxy for health, failing to take into account socio economic status—leading to inequitable outcomes for marginalized populations.
To mitigate these risks, he introduced VAMOS (Vigilant AI Monitoring and Operation System)—a socio-technical framework for real-time AI oversight that integrates governance, team-based monitoring, and analytics to evaluate accuracy, drift, fairness, and equity.
His closing message was both technical and moral: creating AI-enabled learning systems requires trustworthy data, strong governance, and continuous feedback loops. This resonated deeply with the CDISC community’s mission—and with Orizaba Solutions’ commitment to create clarity in data and ensure it is trusted, interoperable, and used responsibly enabling more efficient research and a greater impact on global health.
Plenary: 360i Vision & Roadmap — Accelerating Standards-Driven Automation
The opening plenary, “360i Vision & Roadmap,” captured CDISC’s ambitious journey toward a fully digital and automated clinical trial ecosystem. As clinical research grows increasingly data-intensive—today’s trials collect an average of 3.6 million data points, roughly three times more than a decade ago[1]—the need for smarter, connected data processes has never been greater.
Chris Decker began with a simple but powerful reminder: “AI cannot fix a broken process.” Before we can realize the promise of automation, we must modernize the way research data is structured and shared. The 360i initiative is CDISC’s answer—digitizing study definitions, making metadata interoperable, and enabling machine-driven traceability across the research lifecycle. “CDISC is building models, not monoliths,” Decker explained, emphasizing a modular approach where standards are seamlessly embedded in the tools researchers already use.
Peter Van Reusel showcased the “art of the possible,” illustrating how CDISC standards can support an integrated digital environment from protocol through to submission. Through conceptual models and automation use cases—from protocol optimization to automated eCTD population—he demonstrated how the Unified Study Definition Model (USDM), developed with TransCelerate as part of the Digital Data Flow (DDF) initiative and aligned with ICH M11, can make research faster, more transparent, and more reproducible.
Julie Smiley closed with real-world progress: pilot projects and community collaborations showing that this transformation is already underway. Together, these initiatives are building a foundation for end-to-end automation. This echoed the vision articulated by Dr. Michelle Longmire, CEO of Medable, at an earlier conference on AI in Drug Development (see my previous blog), of the “1:1:1” model: one day to start a study, one day to enroll, and one year to complete.
The 2025 Interchange reinforced why it is imperative that data standards and strategy evolve together. CDISC’s focus on digital transformation, automation, and interoperability mirrors the challenges Orizaba Solutions helps our clients address every day: ensuring that data is high-quality, governed, and ready for AI and analytics.
At Orizaba Solutions, we work with companies, federal agencies, and research organizations to realize the full value of their data. Our mission is to help organizations build trust in their data, modernize processes, and create connected, standards-driven ecosystems that support discovery, compliance, and innovation.
As CDISC enters its next chapter, bridging standards, strategy, intelligent automation, and trustworthy AI, we’re reminded that the future of research depends on one thing above all: enabling data that is accessible, interoperable, and trusted.
Written by Ingeborg Holt, Founder and Principal, Orizaba Solutions
Orizaba Solutions helps organizations unlock the full value of their data through strategy, governance, and data-centric solutions that drive mission success.
[1] Studna, A. Complexities in Data Collection. Applied Clinical Trials, October 2, 2024. https://www.appliedclinicaltrialsonline.com/view/complexities-data-collection
AI in Drug Development: Building Trust, Data Quality, and Collaboration for the Next Era of Innovation
Notes from FDA and Clinical Trials Transformative Initiative (CITTI)’s Hybrid Public Workshop 2025: Artificial Intelligence in Drug & Biological Product Development
Orizaba Solutions believes Artificial intelligence offers unprecedented potential to accelerate discovery, improve clinical trial efficiency, and deepen real-world understanding of therapies. While AI is not a new concept, the rise of large language models (LLMs) marks a transformative leap in what’s possible. Responsible data innovation sits at the heart of healthcare transformation and for AI to reach its promise, the healthcare ecosystem must prioritize data quality, transparency, and collaboration. These themes were front and center at the CITTI–FDA E2025 Hybrid Public Workshop: Artificial Intelligence in Drug and Biological Product Development, where regulators, scientists, and industry leaders explored how AI is reshaping the future of drug development.
The Urgency of Change
The first keynote speaker Dr. Shantanu Nundy, a practicing physician and advisor on artificial intelligence to the FDA Commissioner’s Office, began with a sobering view of U.S. healthcare:
100 million Americans lack regular access to care
Medical error remains the third leading cause of death
U.S. life expectancy now trails other developed nations by four years
Dr. Nundy framed AI as a necessary catalyst to rebuild a healthcare system that too often underdelivers. He highlighted cases showing AI’s growing maturity — from deep learning models that detect structural heart disease better than cardiologists to the potential for AI to reduce animal testing and identify new clinical endpoints and biomarkers.
But his message was clear: the success of AI will hinge not on speed or sophistication, but on trust. As he wrote recently in JAMA, “AI will move drug discovery at the speed of trust.” Democratizing expertise, he argued, must go hand in hand with maintaining accountability and equity.
FDA’s Perspective: Predictability, Transparency, and Context
Dr. Khair ElZarrad, Director of the FDA’s Office of Medical Policy (CDER), also billed as keynote speaker, began with reminding the audience that the FDA is ultimately a consumer of data. With that in mind, he noted 5 guiding principles the agency currently uses to achieve predictability and consistency in regulating AI as applied to medical products:
A risk-based approach to review and oversight
Increased engagements
Transparency
Data governance
Clear context of use
He noted that transparency about what works and what doesn’t is important and will transform this field.
These principles are detailed in FDA’s draft guidance released in January 2025, Considerations for the Use of Artificial Intelligence To Support Regulatory Decision-Making for Drug and Biological Products. Gabriel Innes, Assistant Director for Data Science and AI Policy at OPM, CDER walked through the draft guidance and provided details on the comments. This guidance introduces a risk-based credibility assessment framework — a seven-step process that can be used to establish and evaluate the trustworthiness of an AI model for a particular context of use. The document discusses the importance of life cycle maintenance of the credibility of AI model outputs over time, underscores early engagement with FDA, and emphasizes that documentation and validation should be commensurate with the AI model’s risk and context of use.
Industry Perspectives: From Discovery to Deployment
The first session, Where Are We Now? highlighted how leading organizations are operationalizing AI across the drug development pipeline.
Greg Meyers, Chief Digital and Technology Officer at Bristol Myers Squibb (BMS), described AI as integral to every stage of drug development — from research to manufacturing. In research, AI/ML is being used to inform causal human biology, enabling scientists to infer biological mechanisms from massive, complex datasets. In clinical development, it helps identify disease subtypes, project dosing, and detect biomarkers earlier. He also discussed the potential to use it with Real World Data (RWD) to determine the heterogeneity of a comparator arm population. In manufacturing, AI promises to identify where processes can be optimized.
Thomas Osborne, Chief Medical Officer of Microsoft Federal, framed AI as a tool for “cross-pollinating biomedical knowledge.” He called for breaking silos between laboratory research, clinical trials, and population health. From digital twins that simulate placebo control arms to AI models designing the optimal protocol, Osborne talked about how machine learning and large language models can transform every phase of evidence generation.
Dana Lewis, Patient-Turned Independent Researcher and Founder of OpenAPS, brought a crucial patient voice. Her work using AI to interpret real-world data exemplifies how individuals can identify and fill research gaps — if systems allow it. She urged regulators to create pathways for patient-initiated studies and mechanisms for patients to contribute anonymized health data to research safely. “AI will enable patients to do more with their own data,” she said, “but only if the infrastructure and incentives exist.”
Together, the panel underscored that the next breakthroughs in AI-enabled drug development won’t be technical alone — they’ll be cultural. Collaboration, data sharing, and regulatory clarity must evolve together. The people, the process, the data, the tech that is fit for purpose, all are important components.
Data Quality and the Foundations of Responsible AI
The second session, Data Quality, Reliability, Representativeness, and Access in AI-Driven Drug Development, turned to practice.
Wesley Anderson of the Critical Path Institute reminded attendees that 90% of drug candidates in clinical development still fail and his organization is working to bring down that percentage by bringing together diverse stakeholders to identify blockers, crafting tools for industry use, and working to achieve regulatory success. He noted “Four Pillars” — quality, reliability, representativeness, and access, needed for responsible AI-Driven Drug Development. Without clean, complete, and curated data that reflects real-world diversity and is properly governed, even the best algorithms will perpetuate bias and unreliability.
Anderson highlighted tangible progress, including AI-generated synthetic datasets that mirror real-world populations to support work to qualify a biomarker for clinical trial enrichment in Type 1 Diabetes. His message: the future of AI in drug development is inseparable from the future of data infrastructure.
Michelle Longmire, a physician and CEO of Medable, explored how agentic AI — AI that acts as a collaborative teammate — can relieve operational burdens in clinical research. Her example: an AI “Clinical Research Associate (CRA) agent” that extracts data from multiple systems, identifies site risks, and suggests follow-up actions. By automating the tactical, AI frees humans for the strategic. She also noted during the discussion that “we’ve reached the limit of human potential on drug development- look at the number of drug approvals over time. I am looking to unlock that we haven’t been able to with past technology, older AI and the cloud”. BAM!
Sheraz Khan, Senior Director of Generative AI at Pfizer, discussed efforts to build a shared foundation model for digital health data. Unlike narrow, task-specific algorithms, foundation models can generalize across diverse datasets and devices — a critical step for scalability and fairness. He called for a precompetitive consortium model to develop these tools collectively, ensuring transparency, interpretability, and regulatory alignment.
Moving Forward: AI at the Speed of Trust
The discussions revealed the tremendous potential of AI to accelerate drug and biological product development and reduce costs — but they also underscored the equally important need to advance the standards that ensure safety, efficacy, and quality. Several participants noted that AI is often held to a higher standard than humans. One (non-FDA) speaker noted that society tolerates tens of thousands of traffic fatalities each year, yet a single self-driving car accident can dominate public perception. Whether fair or not, trust in AI — particularly in healthcare — is essential for its adoption. After all, there won’t be just one self-driving vehicle on the road. Like autonomous vehicles, AI in drug development will not exist in isolation; each model, method, and dataset influences the broader ecosystem. Progress, therefore, requires pairing the desire to move rapidly with responsibility for not breaking things when that thing is a human life asking not only what’s possible, but how we can ensure AI is accurate, reliable, and appropriately generalizable. This is possible, when I moved to Austin, TX in 2023, Waymo’s autonomous vehicles were already on the roads there.
Dr. Shantanu Nundy’s framing remains a fitting conclusion: AI will move drug discovery at the speed of trust. That trust must be earned — through data integrity, regulatory collaboration, and a shared belief that technology can serve both science and society.
At Orizaba Solutions, we help organizations build that trust by strengthening the data foundations of innovation. We ensure that AI models, clinical evidence, and regulatory submissions are powered by data that is governed by a process that ensures it is reliable and fit-for-purpose. As the FDA and industry move toward a more AI-augmented future, our mission remains clear: data is the foundation upon which AI algorithms learn, adopt, and generate outcomes, thus the quality and relevance of the data is critical for ensuring accurate, unbiased, and reliable outcomes.
Regulatory Submissions with Real-World Evidence: Successes, Challenges, and Lessons Learned
On September 21st, I attended the 𝗗𝘂𝗸𝗲-𝗠𝗮𝗿𝗴𝗼𝗹𝗶𝘀 𝗜𝗻𝘀𝘁𝗶𝘁𝘂𝘁𝗲 𝗳𝗼𝗿 𝗛𝗲𝗮𝗹𝘁𝗵 𝗣𝗼𝗹𝗶𝗰𝘆 𝗮𝗻𝗱 𝗙𝗗𝗔’𝘀 𝗵𝘆𝗯𝗿𝗶𝗱 𝗽𝘂𝗯𝗹𝗶𝗰 𝗺𝗲𝗲𝘁𝗶𝗻𝗴: 𝘙𝘦𝘨𝘶𝘭𝘢𝘵𝘰𝘳𝘺 𝘚𝘶𝘣𝘮𝘪𝘴𝘴𝘪𝘰𝘯𝘴 𝘸𝘪𝘵𝘩 𝘙𝘦𝘢𝘭-𝘞𝘰𝘳𝘭𝘥 𝘌𝘷𝘪𝘥𝘦𝘯𝘤𝘦: 𝘚𝘶𝘤𝘤𝘦𝘴𝘴𝘦𝘴, 𝘊𝘩𝘢𝘭𝘭𝘦𝘯𝘨𝘦𝘴, 𝘢𝘯𝘥 𝘓𝘦𝘴𝘴𝘰𝘯𝘴 𝘓𝘦𝘢𝘳𝘯𝘦𝘥. The first part of this blog was posted on Orizaba Solution’s linked in page on 9/28, skip to the data and/or afternoon session to see information about the medical products and to hear about the afternoon session.
The meeting opened with remarks from Dr. Sara Brenner, FDA Principal Deputy Commissioner, followed by updates on PDUFA VII and MDUFA V commitments. During the PDUFA VII updates, three main reasons were given for rejecting applications in the 𝗔𝗱𝘃𝗮𝗻𝗰𝗶𝗻𝗴 𝗥𝗲𝗮𝗹-𝗪𝗼𝗿𝗹𝗱 𝗘𝘃𝗶𝗱𝗲𝗻𝗰𝗲 𝗣𝗿𝗼𝗴𝗿𝗮𝗺 (𝗔𝗥𝗪𝗘):
1. Study interpretability concerns
2. Study role in the development program
3. Better suited for established pathway engagement
This resonated with my experience at FDA (2019–2023) when working on data standards for RWE submissions. In the submission reviewed, a randomized controlled study was used for the treatment arm and an RWE study for the comparator. In most of the studies, reviewers had determined the 2 cohorts were not comparable, making analyzing patient-level data for safety and efficacy unnecessary. Despite this, sponsors had often formatted the RWE data into CDISC, but that’s another post😊.
The heart of the meeting was the review of 𝗳𝗼𝘂𝗿 𝗰𝗮𝘀𝗲 𝘀𝘁𝘂𝗱𝗶𝗲𝘀 where RWE was submitted as part of the “adequate and well-controlled study” (AWCS) required for approval under 21 CFR 314.126. (Some may remember Dr. John Concato, former Director of OMP, quoting this section of the CFR so often when referring to RWE questions that he felt compelled to note the agency did not require him to memorize it). Each case study was presented from two perspectives: the sponsor's and the FDA's. FDA presentations focused on 𝘁𝗵𝗿𝗲𝗲 𝗸𝗲𝘆 𝗰𝗼𝗻𝘀𝗶𝗱𝗲𝗿𝗮𝘁𝗶𝗼𝗻𝘀 𝘂𝘀𝗲𝗱 𝗶𝗻 𝘁𝗵𝗲 𝗮𝗽𝗽𝗿𝗼𝗮𝗰𝗵 𝘁𝗼 𝗲𝘃𝗮𝗹𝘂𝗮𝘁𝗶𝗻𝗴 𝗥𝗪𝗘:
🔹Are the RWD fit for use?
🔹Does the study design provide adequate scientific evidence for the regulatory question?
🔹Was the study conducted in line with FDA requirements?
👉 Below are the five takeaways relevant to submission of RWE as part of the AWCS.
𝟱 𝗧𝗮𝗸𝗲𝗮𝘄𝗮𝘆𝘀 𝗳𝗿𝗼𝗺 𝗙𝗗𝗔 𝗖𝗮𝘀𝗲 𝗦𝘁𝘂𝗱𝗶𝗲𝘀
1. Ensure treated and comparator patient cohorts are comparable, especially in non-randomized designs where RWE is used in the comparator arm.
2. Ensure the RWD source provides reliable capture of the primary endpoint for all patients.
3. Select endpoints that are objective and measurable; consider independent adjudication where appropriate.
4. Engage early with FDA on interpretability, endpoints, and statistical approaches.
5. CDER and CBER still requires access to patient-level data for approval of a new drug or indication (CDRH does not)—if unavailable, an established regulatory pathway is likely more appropriate.
Case Studies: Medical Product Details
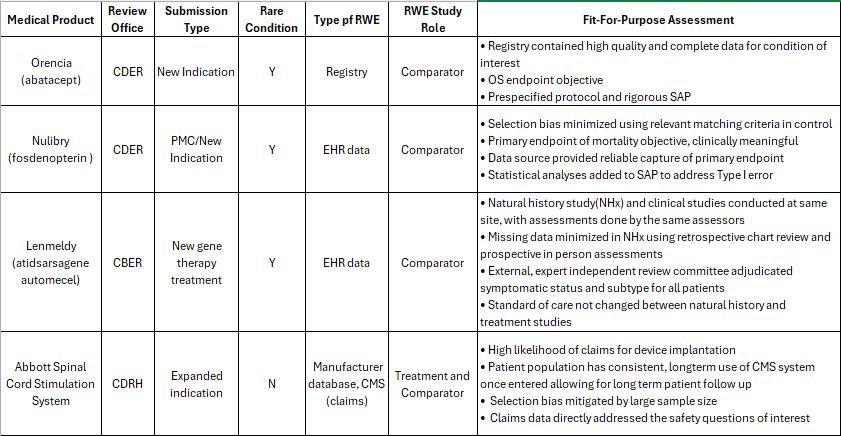
The four medical products using RWE as part of the submission and discussed in the morning sessions.
Exploring the Strengths, Challenges, and Future of Real-World Evidence at FDA
After lunch, Khair ElZarrad, Director of the Office of Medical Policy (OMP), opened the day’s first panel: Strengths and Challenges of Using RWE in Case Studies. This session brought together FDA experts and industry speakers to reflect on lessons learned from the 4 case studies presented at the morning session.
Yun Lu, Deputy Division Director in the Division of Analytics and Benefit-Risk Assessment (DABRA), highlighted several themes from the morning’s case studies:
Three of four applications were for rare diseases.
Registry data played a key role in two cases.
Objective outcomes were selected as primary endpoints.
· Three of the four applications used RWE as external control for a RCT with the treatment.
natural history of diseases, whether used in the approval or not, allowed for an understanding and predictability of disease course important for study design and minimizing bias
Sponsors in all 4 case studies actively addressed bias in the RWE used.
All had early and frequent engagement with FDA and this helped shape their applications
Careful data evaluation, bias minimization, and robust statistical analysis on the part of the sponsors underpinned success.
Motiur Rahman (OMP) added that while sponsors often ask for a “checklist” for RWE, there is no universal template. As former OMP head Dr. John Concato frequently answered when he was asked specific questions about RWD sources, : “it depends.” What matters is whether a study meets the standard of an adequate and well-controlled study (AWCS), as described in 21 CFR 314.126 and this will likely depend on the details of your study design.
When asked about key decision points for using RWE, Dr. Yun emphasized two guiding principles: reliability and relevance.
Reliability means data must be accurate, complete, and traceable.
Relevance means the data must capture critical outcomes and confounders, and include enough patients to power the study.
This explains why a one-size-fits-all checklist doesn’t exist. For example, the CIBMTR registry used in the Orencia approval included every U.S. allogeneic transplant with detailed demographics, making it highly reliable and relevant. In contrast, voluntary registries may be incomplete and unsuitable for certain endpoints. Yun closed with a reminder: RWE must be thoughtfully designed to measure outcomes that truly matter.
Fireside Chat: Envisioning the Future of RWE
The day concluded with a forward-looking discussion moderated by former FDA Commissioner Mark McClellan, now Director of the Duke-Margolis Center for Health Policy. Panelists shared their vision for the next chapter of RWE at FDA:
Dr. Marie Bradley (OMP): Expect more integration and data linkage to create comprehensive datasets and prepare for the growing volume of RWE trials.
Dr. Mallika Mundkur (Office of the Commissioner): RWE and AI remain top priorities, especially through collaboration with NIH, CMS, and other agencies.
Dr. Shantanu Nundy (FDA AI Advisor): Shared a personal story illustrating the value of registries in clinical decision-making. He noted that using AI to look across hospital records to answer questions both clinicians and patients might have would be useful. I would argue putting money into registries is likely to yield better results than AI, at least based on the evidence we have so far.
Dr. Daniel Caños (CDRH): Highlighted the promise of unique device identifiers to connect data sources and improve evidence for devices.
Panelists acknowledged ongoing challenges—interoperability, quality, and traceability—but expressed optimism. They pointed to new initiatives such as Sentinel 3.0 and CDRH’s collaboration on the National Evaluation System for Health Technology (NEST) as steps toward stronger evidence generation and better regulatory decision-making.
Takeaway:
The discussions underscored both the progress and complexity of integrating real-world evidence into regulatory science. While there’s no single roadmap, the principles of reliability, relevance, and early FDA engagement continue to guide successful RWE applications. Looking ahead, greater data connectivity, insight and analysis from cross-agency collaborations, and innovative infrastructure projects promise to expand the role of RWE in shaping the future of drug and device development.
Optimizing Real-World Evidence Use at FDA, Workshop Insights
December 18, 2024
Introduction
Improved access to digital healthcare data of all types and the ability to rapidly analyze data has led to interest in the unique insights this data might yield to help us all live healthier lives. Real-world data (RWD) and the real-world evidence (RWE) derived from it have the potential to transform drug development, promising to bring down the cost and increase the diversity of study populations. As the volume and variability of RWD continues to grow, important issues regarding data quality, determining fit-for-use, and appropriate methods to analyze non-randomized observational studies where patients are selected from large datasets exist. With more than 131 million people, 66 percent of all adults in the United States, using prescription drugs and with utilization particularly high for vulnerable groups, including older adults and those with chronic conditions[1], the FDA must take the time to understand these issues before RWE can become a regular part of submissions and regulatory decision making.[2] A recent workshop hosted by the Duke-Margolis Health Policy Center brought together representatives from the FDA, industry, academia, and non-profit organizations to discuss the current and future activities of the FDA's RWE Program for drugs and biological products. Participants explored the opportunities and challenges of using RWD and RWE in regulatory decision-making and how to build on the accomplishments and knowledge gained in the 6 years since FDA published their Framework for FDA’s Real-World Evidence Program in 2018. FDA is actively seeking input to help facilitate the future direction of the RWE Program, see FDA Docket: FDA-2024-N-5057 for more information, the comment period ends 1/13/2025.
Center for Clinical Real-World Data Integration (CCRI)
This workshop covered a lot of ground and generated a lot of discussion and insights covered below. The big announcement at this meeting came when the third speaker, Dr. Corrigan-Curay, formerly CDER’s Office of Medical Policy Director and now the Principal Deputy Center Director, began her RWE Future Directions session with the announcement of the new Center for Clinical Real-World Data Integration (CCRI). The center has been created to assist with the increasing volume and complexity of regulatory submissions involving RWD and the use of emerging technologies such as artificial intelligence (AI) and digital health tools (DHTs) to collect, organize, and analyze RWD. CCRI will focus on the following:
· Scientific Review and Policy
· Coordinated Outreach and Engagement
· Research Initiatives
· Knowledge Management, including training
Agenda Highlights
Commissioner Califf opened the meeting remotely from California, where he self-reported tromping around pastures engaged in work on the avian influenza outbreak in cows. He expressed his enthusiasm for RWD and RWE, and its importance for the future of FDA and for therapeutics and diagnostics. Dr. John Concato, Associate Director for Real-World Evidence Analytics at CDER, took the stage next and provided an overview of FDA’s RWE Program followed by Dr. Corrigan-Curay’s insights on RWE’s future, including the announcement of the new CCRI. She was then joined on stage by Dr. Concato for a fireside chat. After a break, there were two panel sessions Opportunities and Challenges When Using Real-World Data, and the second panel and last session: Methodological Considerations for Real-World Evidence. Panelists in both sessions were from FDA, industry, academia, and nonprofits.
Overview of FDA’s RWE Program
John Concato, Associate Director for Real-World Evidence (RWE) Analytics in the Office of Medical Policy (OMP), began his session with a historical overview of RWD, starting in the 1990s.[3] He described how medical students and epidemiologists were traditionally taught using the "pyramid of strength of evidence," (see Fig. 1) which places randomized

controlled trials (RCTs) at the top and observational studies lower down, but not the lowest. This pyramid, though foundational, is an oversimplification, Dr. Concato noted, and that while RCTs are more trustworthy, the interpretation that non-randomized studies are untrustworthy is incorrect. This blog author also wants to point out that observational studies still rank higher than anecdotes and personal opinions, which are the weakest level of evidence. The origins of the pyramid date back to 1976 when The Canadian Task Force on the Periodic Health Examination was established to determine how the periodic health examination might enhance or protect the health of the population.[4] This may be the topic of a future blog post.!
Figure 1: Strength of Evidence Pyramid
Dr. Concato highlighted debates in peer-reviewed literature over the merits of real-world data (RWD), including the Hormone Replacement Therapy (HRT) study, where differences in analysis impacted coronary heart disease risk evaluations. His key takeaway: getting RWE right is challenging but achievable.
Two recent approvals based on RWE exemplify its potential. A new indication for Prograf was approved based solely on a non-interventional study, and another approval for a dose regimen change for Vimpat also used RWE data. Both applications met the FDA’s 21 CFR 314.126 requirement for “adequate and well-controlled studies” with validity comparable to RCTs. Dr. Concato noted, as he often does when discussing RWE, that FDA does not require him to memorize this regulation, he just repeats it so often that he has memorized it.
Prograf’s approval involved several factors that contributed to the successful use of RWE:
The source data was a registry covering all lung transplants during a specified period.
An observational design comparing treatment to historical controls, with an analysis plan and patient-level data provided to the FDA.
The outcomes used were organ rejection and death, where the dramatic effect of treatment minimized bias as an explanation.
Dr. Concato addressed challenges in using RWE, in the areas of source data fitness for use, study design and interpretation (e.g., confounding and immortal time bias), and proper study conduct, including lack of access to patient-level data. He emphasized that RWD and RWE are not new concepts; rather, electronic access to clinical data is evolving, making it more relevant and reliable. Next, he reviewed FDA’s extensive RWE-related accomplishments, such as guidance documents, demonstration projects on data, study design, and tools, and external collaborations, including those with ICH and EMA., noting that these efforts satisfy 21st Century Cures Act and PDUFA mandates. Dr. Concato concluded by citing a 2022 NEJM article he co-authored, reaffirming FDA’s commitment to robust policy development and maintaining evidentiary standards to protect and promote public health. The progress of the FDA’s RWE program underscores this dedication.
RWE Future Directions
Dr. Corrigan-Curay began her session with the announcement of the new Center for Clinical Real-World Data Integration (CCRI), as noted above. The Center will help CDER create a more centralized structure to help define strategic objectives, promote consistent scientific review and knowledge management, coordinate research goals, and engage the external real-world evidence (RWE) ecosystem. Its vision is to optimize the use of RWD when used to inform the effectiveness and safety of drugs and biological products, and its mission is to advance the evaluation of RWD by addressing gaps in regulatory science and policy related to RWE and by communicating CDER's priorities regarding using RWE in support of pre- and post-market regulatory decision-making in CDER.
Drs Concato and Dr. Corrigan-Curay answered questions during the Fireside Chat with FDA Leadership. John Concato mentioned his planned retirement, announced well before the election, he made sure to clarify. Dr. Corrigan-Curay did hint that he may be involved in a part-time capacity, but it remains to be seen if the 21 CFR 314.126 will keep its pole position in Dr. Concato’s memory or be replaced with information from other pursuits.
After a break, there were two panel sessions, which allowed panelists to give 5-minute opening remarks and then answer questions from the panel moderator and the audience. I have summarized the remarks and key points of discussion below.
Panel Session 1: Opportunities and Challenges When Using Real-World Data
Dr. Tala Fakhouri, the Associate Director for Data Science and Artificial Intelligence in the Office of Medical Policy at CDER, highlighted the transformative potential of artificial intelligence (AI) in accelerating drug development across all phases, from preclinical research to post-market surveillance. She emphasized that AI can be a powerful tool for generating evidence from RWD, but its success depends on the same prerequisites as RWD itself: high-quality, fit-for-purpose data. Dr. Fakhouri pointed out the need for “multi-data fluency” within organizations, urging greater collaboration between data scientists and domain experts—a challenge bioinformatics successfully tackled in the (now historic) 1990s, so maybe some lessons to be learned there.
Donna Rivera, the Associate Director for Pharmacoepidemiology in the Oncology Center of Excellence (OCE) at FDA, shared insights from the OCE, which has conducted over 200 consultations involving RWD applications. She emphasized the importance of ensuring data quality, particularly in pragmatic clinical trials, and stressed the methodological rigor required to enable RWD to yield insights that provide patient benefit. Dr. Rivera also underscored the work being done to Understand how RWD can be applied for regulatory purposes and the potential for RWD to modernize evidence generation across the agency.
Jingyu (Julia) Luan, Executive Regulatory Science Director at AstraZeneca (AZ), emphasized that data quality is the bedrock of their work. She referenced AZ’s DAPA-MI trial, which demonstrated the feasibility of registry-based randomized controlled trials (R-RCTs). This innovative approach accelerated trial recruitment, reduced costs, and minimized patient site visits, showcasing the efficiency gains possible with high-quality RWD.
Nicole Mahoney, an Executive Director for Regulatory Policy at Novartis, discussed creating workflows to ensure data reliability and relevance within the industry. She raised the practical challenges of assessing vendors and measuring data quality for regulatory purposes. She also touched on the complexities of harmonizing definitions and guidance across international regulatory bodies, a task that likely falls outside the FDA’s remit but could be addressed by collaborative initiatives, perhaps a role for TransCelerate?
Dan Riskin, MD, MBA, Founder and CEO of Verantos and a Clinical Professor at Stanford University, pointed out the potential safety implications of using electronic health record (EHR) data in learning health systems if the underlying data is not accurate. Drawing on findings from the TRUST study, his findings show that data quality can significantly influence patient cohort definitions and study outcomes. This blog author’s takeaway: there is a good reason that trials have historically been done outside the healthcare system. Riskin advocated for further research into the implications of data quality.
Themes Explored and Key Takeaways
Data Quality: Panelists universally agreed on the importance of reliable, relevant, and high-quality data. However, there is no consensus yet on how to systematically improve RWD quality or establish robust reporting standards for reporting on data quality.
Role of AI: AI’s potential to enhance RWD analysis was a central topic. Discussions included creating datasets for AI testing, ensuring algorithm generalizability, and addressing the dependence of generalizability on data relevance.
Regulatory Considerations: There are highly varied capabilities in creating datasets; some extract data well, including verification and traceability, but others don’t. The panel explored how to maximize RWD usefulness while maintaining scientific rigor.
Context Dependence: There are no hard and fast rules for RWD, it depends a lot on the question or questions being asked of the data. What to measure in the data to prove that it is fit for use and of high enough quality for regulatory purposes is use-dependent.
Collaborative Approaches: Bridging the gap between data scientists, engineers, and domain experts is essential for the effective use of RWD and creating the right analysis tools.
Registries as a Solution: Patient registries could play a pivotal role in improving data availability and reliability, as seen in the development of AstraZeneca’s DAPA-MI trial. (This blog author agrees and recommends interested readers investigate the role of a registry in developing treatments for cystic fibrosis.)
Methodological Considerations for Real-World Evidence
Jennie Li, PhD and Associate Director for RWE at the Office of Surveillance and Epidemiology in CDER, highlighted four pivotal efforts at the FDA to advance RWE methodologies: New tools and methods for pulling data from EHRs and the use of data to pull more reliable cohorts, the Use of Sentinel to estimate the potential impact of confounding and bias in RWD, a framework and tools to identify patterns of missingness and work on a causal inference framework. Ms. Li emphasized key questions researchers should address when designing RWE studies, such as selecting appropriate endpoints, assessment frequency, what comparators can be used if the standard of care is not measurable and understanding disease progression and known pragmatic factors.
Yun Lu, PhD and Deputy Division Director at the Division of Analytics and Benefit-Risk Assessment in CBER’s of Biostatistics and Pharmacovigilance, provided insights into the unique challenges of vaccine research in RWD. Vaccines are administered to healthy populations and may introduce biases as vaccine recipients may inherently be healthier than people who chose not to be vaccinated. To address these biases, the FDA is developing statistical methods to reduce unmeasured confounding in vaccine effectiveness studies.
Simon Dagenais, PhD and Lead for Methods & Innovation in the RWE Platform at Pfizer, advocated for hybrid RCT/RWE approaches in chronic disease research, such as Type 2 Diabetes (T2D) where the disease and endpoints are well characterized and where pre-market approval studies often require 8-10K patients, take over 6 years, and cost a lot of money. His suggestions included utilizing augmented controls derived from RWD and implementing a mix of passive follow-up and scheduled visits to enhance study efficiency.
Stefan James, Professor of Cardiology and Scientific Director of Uppsala Clinical Research Center at Uppsala University, highlighted the role national registries can play. He suggested national registries with defined core variables and federated data access could facilitate analyses. He also noted that complex study outcomes would likely need some level of adjudication.
Sebastian Schneeweiss, M.D., Sc.D., and Professor of Medicine at Harvard Medical School (among other positions), underscored the need for rigorous benchmarking and data quality assessment, a theme heard in session 1. He built on Dr. Dagenais’s use case and described a possible way to expand effectiveness working with RWD, including first benchmarking a drug’s early RWD findings against the phase 3 trial results to validate data reliability and then expanding from there, stepwise, into other populations and/or clinical endpoints. He also addressed data quality, noting that many would be surprised at how poor their data quality is if they did measure it. However, understanding the flaws is a necessary step for improving the quality.
Themes Explored and Key Takeaways
Endpoints and Validation: While overall survival (OS) remains a straightforward endpoint, more complex endpoints require validation and/or adjudication and need to consider the disease trajectory, treatment landscape, and study context.
Data Quality and Reporting: Robust RWE requires high-quality data. Researchers should focus on describing datasets, defining variables, and setting quality benchmarks to enhance validity and reproducibility.
Understanding measurement imperfections and aligning data sources with desired characteristics are important for ensuring study results are meaningful.
Sharing Lessons Learned: Given the complexity and cost of validating RWD, panelists emphasized the importance of sharing best practices and lessons learned. Not every sponsor should have to analyze a dataset to understand the quality, data vendors could provide this information.
Frameworks and Methodologies: Sentinel’s advancements in handling missing data, ongoing work to understand causal inference, and exploration of confounding and bias in linked versus unlinked datasets will provide valuable tools for addressing common RWE challenges.
Pragmatic Approaches/ Innovative Data Integration: Combining structured and unstructured data, using the former for cohort selection and the latter for subsequent characterization of other factors.
Methods blending RWD with traditional RCT data to create adaptable and efficient study designs.
Conclusion
Real-world evidence has a pivotal role in the future of regulatory science. The FDA is continuing its commitment to supporting the use of RWE in both premarket approvals and post-market surveillance through research, demonstration projects, and initiatives like CCRI. There are many opportunities for innovation with RWD, RWE, and AI that can both help to improve data quality and analyze data. However, fitness for use, data quality metrics, and appropriate analysis methods are still evolving and need to be better understood before the information can be used without close human supervision and analyses. The FDA has not changed its data standards[5] and will continue to require “[a]dequate and well-controlled studies” for regulatory decision-making (see 21 CFR 314.126) whether the data comes from randomized clinical trials, observational studies, pragmatic trials, or another design.
Progress in RWE requires a unified effort from all stakeholders: regulators, researchers, industry, physicians, patients, payers, and EHR vendors. The FDA is interested in hearing from all stakeholders about their experience with RWD and RWE and how the FDA can best move forward to use this data to improve the health of the public. Please comment on FDA Docket: FDA-2024-N-5057.
A big thanks to Duke Margolis, FDA, the speakers, organizers, and participants for an interesting and thought-provoking meeting. I look forward to updates from the CCRI and continued progress on the use of RWD and RWE to help patients live healthier lives.
Footnotes
[1] https://hpi.georgetown.edu/rxdrugs/
[2] Note that RWD has long been used by sponsors to inform study design. Also, FDA has used submitted RWE in regulatory decision-making.
[3] Yes, it is hard to swallow but the 1990s is now considered historic.
[4] Canadian Task Force on the Periodic Health Examination (3 November 1979). "Task Force Report: The periodic health examination". Can Med Assoc J. 121 (9): 1193–1254. PMC 1704686. PMID 115569
[5] In this context, I am using standards to mean “quality” or “grade”, not “the rules that define how data is structured, described, and shared”. It is worth noting that FDA has not changed their data standards for submission of RWE, CDISC SDTM and ADaM are still required, but they are actively exploring whether a change is needed .

Digital Health Technologies and Electronic Health Records: Power and Problems
Digital health technologies and electronic health records are revolutionizing the healthcare sector and the future of healthcare is undeniably digital.
Challenges still exist, but work is being done to solve them. As technology continues to evolve and improve, we can expect even greater advancements in digital health tools, EHRs, and the use and analysis of the data collected from both technologies. Healthcare providers and patients must work together to ensure that accurate data is being collected and used in ways that facilitate its use in identifying optimal healthcare, both for the patient and for the wider US population.
September 6, 2024
by Grete McCoy MPH, RDN, CDCES
Introduction
Digital health technologies (DHTs) and Electronic Health Records (EHRs) have revolutionized today's healthcare. Efforts to develop EHRs began in the 1960s and ’70s, when academic medical centers developed their own systems[1]. The passing of the Health Information Technology for Economic and Clinical Health (HITECH) Act in the US in 2009 spurred rapid adoption of EHRs in the United States with over 80% of hospitals using EHRs in 2015 compared to less than 10% in 2008.[2] EHRs have also evolved rapidly and now streamline scheduling, handle referrals, take care of billing, send prescriptions to pharmacies and order durable medical equipment (DME). They have facilitated increased access to healthcare providers (HCPs), and, with the help of DHTs, provide HCPs access to real-time data for faster response to issues like blood sugar levels and blood pressure. However, EHRs and DHTs are not without challenges. As a registered dietitian treating diabetes since the mid-1990s, I’ve had a front-row seat in seeing how this technology is reshaping healthcare.
Outside the Clinic Walls: Digital Health
Digital health technology can refer to digital technology for a wide spectrum of health-related metrics, but the FDA defines a DHT as a system that uses computing platforms, connectivity, software, and/or sensors, for health care and related uses[1]. DHTs are playing a growing role in health care, more offices are utilizing remote monitoring to stay in touch with patients between visits. Wearable Devices, like continuous glucose monitors, heart monitors, and smartwatches provide real-time data on health metrics such as blood glucose to heart rate to sleep patterns to physical activity. This data helps both patients and HCPs to manage health conditions, medication effectiveness and adherence, and even lifestyle choices. A typical physician visit for someone with diabetes is every three months, and labs to assess treatment are only drawn at those visits. With remote monitoring, an HCP can log in to a designated portal, review their patients’ real-time blood glucose or blood pressure, assess medication and activity levels, and, if needed, make medication adjustments. It can be frustrating for both physician and patient if something has not been working and it is not addressed until the patient’s next scheduled visit, 3 months later. When using a wearable device, the HCP reviewing the data must know their patient and the standards of care for the condition being monitored. In addition, they need to know the device, how it works, and the data it provides. Patients also require training to be able to use the device correctly and link to the HCP system.
Inside the Clinic: Electronic Health Records (EHRs)
Healthcare providers are mixed in their views of EHRs with some disliking the constrained data entry, often requiring the use of drop-down lists, and discouraging (or prohibiting) the use of written notes. However, they also appreciate the ease of access to patient data, including labs and imaging data, from outside the office. EHRs facilitate cutting and pasting and if data has been entered incorrectly it can be carried through to subsequent visits without anyone checking. Once while reviewing working at a pediatric hospital, I noticed the age of the patient was recorded as 48. Going back through the notes from 6 office visits, I eventually found the correct age of 4 years and 8 months. Other drawbacks include power or internet outages and data that was not saved correctly (or at all).
Ideally, EHRs should centralize patient data, making it easily accessible to multiple authorized healthcare providers. Patient medical history, lab results, and treatment plans should be available at the point of care, to improve both efficiency and accuracy. Unfortunately, not all systems are interoperable, and although the newly renamed Assistant Secretary for Technology Policy/Office of the National Coordinator for Health IT (ASTP/ONC) is working to change this, it is often left to the patient to make sure a specialist has all the information needed for a consult. The adaptation of Patient Portals alleviates some of these issues, giving the patient access to their records and the ability to send them to anyone they choose.
Data
EHRs collect a vast amount of data that can be analyzed to identify trends, improve treatment protocols, and enhance patient outcomes. Data analysis helps in identifying at-risk populations, incidence of chronic conditions, or tracking the effectiveness of new treatments.[2] They can also be used to track the performance or level of care a physician provides, however, caution should be used as information can be entered and/or interpreted incorrectly. The extreme detail in the ICD10 coding can lead to confusion over the best coding choice to enter and there can be a discrepancy between the true diagnosis and the code entered. For example, a new patient who did not bring past lab work might be misdiagnosed as having acute renal failure when they have CKD (chronic kidney disease). A diabetes patient seen via a telemedicine visit might tell a primary care provider (PCP) their A1c has been good, and they have no additional problems leading to a code for “type 2 diabetes without complications” when a physical exam would have shown poor pedal pulses and a random blood sugar might be elevated indicating the correct diagnosis is “type 2 diabetes with peripheral circulatory complications uncontrolled”. Confounding that problem, a skilled biller/coder knows that some codes will not be reimbursed or are reimbursed at a lower level than others, and codes that provide reimbursement may be used preferentially. For example, CKD is reimbursed where Kidney Disease Stage 3 is not, and the HCP may choose to code CKD and note the stage in the narrative. This can cause problems or at least extra work for studies that are seeking to use a stage or the disease as inclusion or exclusion criteria for an RWD clinical trial. These differences in coding and subsequent reimbursement can be a large factor between a thriving small practice and having to shut down.
Medication switching trends may also be misinterpreted due to a PCP having to change medications to those that will be covered by a patient's health plan, not because the initial medication was ineffective. When this happens, the reason for the switch is rarely documented in the EHR. Users of EHR data for secondary purposes, such as supporting safety or efficacy for a placebo arm of a new drug or biologic, need to be aware of potential errors in EHRs as well as regional practices in coding and standard of care which could bias results especially when pooling data from patients in different areas and with diverse demographic profiles.
Patient Engagement
Many EHR systems include patient portals to allow individuals to access their health information, schedule appointments, and communicate with their healthcare providers. This increased transparency and access will ideally lead to increased patient engagement and that should lead to better health outcomes and patient satisfaction. However, HCPs and allied health practitioners may decide to not document critical information such as patient comprehension, adherence to treatment, and other issues in the EHR if it could upset a patient. This is especially a concern for pediatric or geriatric practices where parent or caretaker behavior and attitudes can impact treatment. An office doesn’t want to risk a caregiver not bringing the child or patient in for care. Technology can be used to address this but would need to comply with rules addressing patient data access.
Moving Forward
Digital health technologies and electronic health records are revolutionizing the healthcare sector and the future of healthcare is undeniably digital. Challenges still exist, but work is being done to solve them. As technology continues to evolve and improve, we can expect even greater advancements in digital health tools, EHRs, and the use and analysis of the data collected from both technologies. Healthcare providers and patients must work together to ensure that accurate data is being collected and used in ways that facilitate its use in identifying optimal healthcare, both for the patient and for the wider US population. Innovations for secure data sharing, increased usage of linked and wearable technology, and more sophisticated algorithms for data checking as well as for analysis and diagnosis offer multiple opportunities for increasing healthcare access, improving healthcare delivery, empowering patients to participate in their care with the potential to contribute to improved patient outcomes and a healthier population.
[1] Digital Health Technologies for Remote Data Acquisition in Clinical Investigations Guidance for Industry, Investigators, and Other Stakeholders, December 2023.
[2] Ryan, D.H., Lingvay, I., Deanfield, J. et al. Long-term weight loss effects of semaglutide in obesity without diabetes in the SELECT trial. Nat Med 30, 2049–2057 (2024). https://doi.org/10.1038/s41591-024-02996-7
[1] Atherton, Jim. Development of the Electronic Health Record, https://journalofethics.ama-assn.org/article/development-electronic-health-record/2011-03
[2] Ratwani R. Electronic Health Records and Improved Patient Care: Opportunities for Applied Psychology. Curr Dir Psychol Sci. 2017 Aug;26(4):359-365. doi: 10.1177/0963721417700691. PMID: 28808359; PMCID: PMC5553914.

5 Takeaways & An Overview of FDA's Guidance on Assessing Electronic Health Records and Medical Claims Data for Regulatory Decision-Making
FDA released their updated and final guidance on Assessing Electronic Health Records and Medical Claims Data to Support Regulatory Decision-Making on July 25, 2024. The guidance does not endorse specific data sources, study methodologies, or standards. Instead, it provides insight into the FDA’s concerns and considerations so sponsors can address these when conducting studies using data from EHRs and medical claims.
5 Takeaways & An Overview of FDA's Guidance on Assessing Electronic Health Records and Medical Claims Data for Regulatory Decision-Making
Introduction
FDA released the final guidance, Assessing Electronic Health Records and Medical Claims Data to Support Regulatory Decision-Making, on July 25, 2024. This work follows the Framework for FDA’s Real-World Evidence (RWE) Program released in 2018 and 5 additional guidances concerning Real-World Data (RWD) released subsequently in the wake of the 2016 21st Century Cures Act which called for the FDA to explore the potential of RWD in regulatory decision-making. Three centers within the FDA developed the guidance: the Center for Drug Evaluation and Research (CDER), the Center for Biologics Evaluation and Research (CBER), and the Oncology Center of Excellence (OCE), to clarify the agency’s thinking on the use of RWD coming electronic health records (EHRs) and medical claims in clinical studies to support new drug and biologic approvals.
Background
FDA has used RWD for post-market surveillance for some time and RWD has been used sparingly for new drug approvals and expanded indications. The agency does not require randomized clinical trials (RTCs) to support drug approval, it requires “adequate and well-controlled clinical investigation” as codified in Code of Federal Regulations (CRF), Title 21, Chapter I, Subchapter D, Part 314. OMP’s Dr. John Concato, speaking at a Regan Udall RWD/RWE Guidance Webinar Series meeting, noted that FDA staff aren’t required to memorize this section of the federal code but, refer to it so often it becomes part of memory.
About the Guidance
The guidance spans 39 pages and covers a broad range of considerations, too extensive to be addressed in a single blog post. Some of the content mirrors discussions from FDA’s 2013 Guidance for Industry and FDA Staff: Best Practices for Conducting and Reporting Pharmacoepidemiologic Safety Studies Using Electronic Healthcare Data, which was developed to fulfill Prescription Drug User Fee Act IV (PDUFA IV) commitments.
FDA does not endorse any specific data sources, study methodologies, or data standards in this guidance. Instead, it offers considerations for conducting adequate and well-controlled clinical investigations using data from EHRs and claims data. Key areas of focus include data sources, data capture, missing data, and validation.
The guidance starts by noting that sponsors should submit protocols and statistical analysis plans before conducting an RWD study. All essential elements of study design, analysis, conduct, and reporting should be predefined as required RCTs. The FDA isn’t reinventing the wheel with RWD but adapting it so the ride will be safe and effective on a broader range of terrains.
Data Sources
Given the current variability of RWD, including EHRs and claims, each data source proposed for a study needs to be evaluated within the context of the study question. The appropriateness of a data source for a particular study depends on its containing the right data elements over a sufficient length of time to answer the study question. For example, understanding whether a data source from a given health system comprehensively captures a patient’s care is necessary to determine whether the source includes all relevant outcomes.
Key Study Variables
The selection, definition, and validation of key study variables are critical elements of a RWD study. Key study variables include study population inclusion and exclusion criteria, exposure, outcome, and covariates. The guidance proposes 2 sets of definitions be developed for key study variables, a conceptual definition, and an operational definition. The conceptual definition “should reflect current medical and scientific thinking regarding the variable of interest”. The operational definition consists of the data element(s) and value(s) in the RWD used to determine if the conceptual definition is satisfied. It is recommended that all definitions be included in the study protocol and that the potential misclassification of all key study variables be evaluated along with the possible impact on study findings.
Missing data is a common challenge in the use of RWD and the extent of missingness for key variables should be assessed. It is important to understand if the missing data was intended to be collected in the source data or if the data elements are absent because they are not typically collected in the type of real world data being used. For example, blood pressure isn’t collected in medical claims, but a claims code can indicate high blood pressure. Sponsors need to understand the reasons for the absence of information in each RWD source and evaluate if it is it missing at random or if it could introduce bias into the study. The understanding and assumptions around missing data should be reflected in the study protocol and the statistical analysis plan.
Study Design Elements
The FDA is aware of the risk of multiple testing in the data by the sponsor with only the data yielding the desired results being submitted. The guidance notes that “[t]he study questions of interest should be established first, and then the data source and study design most appropriate for addressing these questions should be determined.” However, the boundary between analysis to determine if a data source is fit for purpose and conducting the study itself is still a bit blurry.
Changes in standard of care, treatment methods, coding conventions, and coding versions over time can affect study results. The guidance notes the importance of considering the time period from which subjects are selected from an RWD source to understand any potential impacts on the fitness of the data source for the study.
Data Quality & the RWD Lifecycle
RWD used in clinical studies undergoes several stages, starting with data accrual in the source. A subset of that source data will be extracted, standardized, and then stored for use. The FDA refers to this process as curation. The curated data may then be converted into another format or structure (defined by FDA as transformation) and, if needed, de-identified before being used in a study-specific dataset for analysis. The guidance specifies that sponsors should validate that there is no loss of information in this process. However, the FDA does not currently endorse any set of guidelines. Some may remember a 3rd party initially created the original CDISC data checks before the FDA took control of the work. A similar process may occur for RWD.
The guidance includes the expectation of documentation of the data management process and quality management for the lifecycle of the RWD as it moves to becoming RWE. This documentation should consist of a QA/QC Plan. These data and quality management processes are established for RCTs but have yet to be standardized for RWD, something that will likely evolve with time and as the FDA and sponsors gain more experience with RWD.
Conclusion
This guidance marks a step forward in integrating EHR and claims data into the regulatory framework of drug and biologic approvals. While it provides important considerations for conducting RWD studies, the guidance underscores the importance of maintaining the basic elements of good clinical practice. By addressing considerations around the use and analysis of EHR and claims data to support a clinical study, this guidance provides a set of factors that can be used to evaluate RWD data sources to conduct studies that can be effectively and reliably used to support regulatory decisions. By taking this approach instead of being prescriptive, the agency leaves room for more exploration and innovation with RWD. As the use of RWD in clinical studies continues to evolve, ongoing dialogue between the FDA, sponsors, and other stakeholders will be essential in refining these processes and ensuring the safety and efficacy of new therapies.
5 Key Takeaways
1. The standard of adequate and well-controlled clinical investigations needed for drug approval (CFR 21 Part 314) also applies to RWD studies.
2. The guidance does not endorse specific data sources, study methodologies, or standards. Instead, it provides insight into the FDA’s concerns and considerations so sponsors can address these when conducting studies using data from EHRs and medical claims. This is a balance between FDA ensuring studies conducted in RWD can be evaluated for drug approval without stifling innovation in the use of this data by being overly prescriptive.
3. Pre-specification is important. The balance of how much investigation can be done in the source data to determine fit for purpose before defining key study variables is still undefined. The FDA is aware of the risk of multiple testing by the sponsor with only the results yielding data set being submitted.
4. The guidance proposes 2 sets of definitions be developed for key study variables, a conceptual definition, and an operational definition and these be documented in the protocol. Key study variables include study population inclusion and exclusion criteria, exposure, outcome, and covariates.
5. RWD should be characterized for the completeness, conformance, and plausibility of data values. Work is required to ensure RWD is not altered meaningfully from the source data to the final data set analyzed and ultimately submitted to the FDA. The submission of documentation of the Data Management Process and Quality Management, including documentation of the Quality Assurance (QA) and Quality Control (QC) Plan.